Introduction
***TESTICULAR TORSION IS A SURGICAL EMERGENCY***
Many boys and men have lost their lives due to late diagnosis and treatment of this condition. It is not the most common cause of acute scrotum but is the most important as late management results in irreversible ischemia and gonadal necrosis. Lack of awareness and late presentation by patients and parents also contribute to delayed management of this condition.
Epidemiology
Testicular torsion may be divided to intratunical or intravaginal torsion and extratunical or extravaginal. This refers to the torsion occurring with the tunica vaginalis invaginating the testes or not.
Extratunical torsion occurs mostly during the perinatal period as the loose areolar tissue surrounding the testes predispose to torsion during descent. 70% occurs prenatally, while 30% occurs postnatally. Adherence of the testes to the surrounding tissues usually occurs around 6 weeks of age.
Intratunical torsion mostly occur with the puberty age group of boys (13 - 16 years). This might be related to testicular enlargement due to increased testesterone production during this period. The left testes is more frequently involved.
This mostly occur in the bell-clapper anomaly type of testes when the tunica vaginalis invaginate superiorly involving the spermatic cord, instead of just the epididymis and testes. In the normal testes, the posterior part of the testes is anchored to the scrotum preventing from much mobility. However when completely surrounded by the tunica vaginalis, the testes becomes more mobile at its axis through the spermatic cord. This anomaly usually occurs bilaterally requiring contralateral orchidopexy during surgery.
 |
| Bell-clapper: 5. Tunica vaginalis. 9. Testes. |
 |
| Angular relations between the testes and the tunica vaginalis. Testes lies more horizontal with higher invagination of the tunica. |
Cryptochordism is also associated with higher incidence of torsion. Trauma and exercise causing higher cremesteric activity may also result in torsed testes.
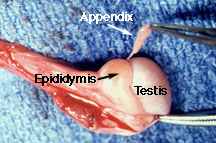
The commoner but less serious torsion of the testicular appendix (Hydatid of Morgagni) occurs also most often at puberty. The appendage is present in 90% of boys. In a review of 771 children with acute scrotum, 58% had torsion of the testicular appendix, 29% of the testes.
Rarely, the testes is torsed between it and the epididymis connected loosely.
Clinical Features
***Epididymo-orchitis is rare in children and adolescents***
The typical feature is sudden onset of pain at the scrotum or the ipsilateral iliac fossa. This may or may not be accompanied by nausea and vomiting. Beware that some may present with gradual onset of pain leading to delayed diagnosis. Resolving pain may indicate spontaneous resolution of the torsion or worse, a necrotic testes.
Examination reveals inflammed testes which is tender to touch. The testes is usually high riding as well with absence of the cremasteric reflex. However absence of reflex should not be the only indicator as there are case reports of presence of this reflex in a case of testicular torsion. The presence of the bell-clapper anomaly makes the diagnosis more likely and indicated for surgical exploration. Secondary reactive hydrocele may make testicular palpation more difficult.
In a torsed testicular appendix, the pain is usually not as severe and more localized. A 'blue-dot sign' may be seen and point palpation is tender, while palpation of the whole testes is not. However, this is difficult to elicit once secondary hydrocele develop and the scrotum becomes edematous.
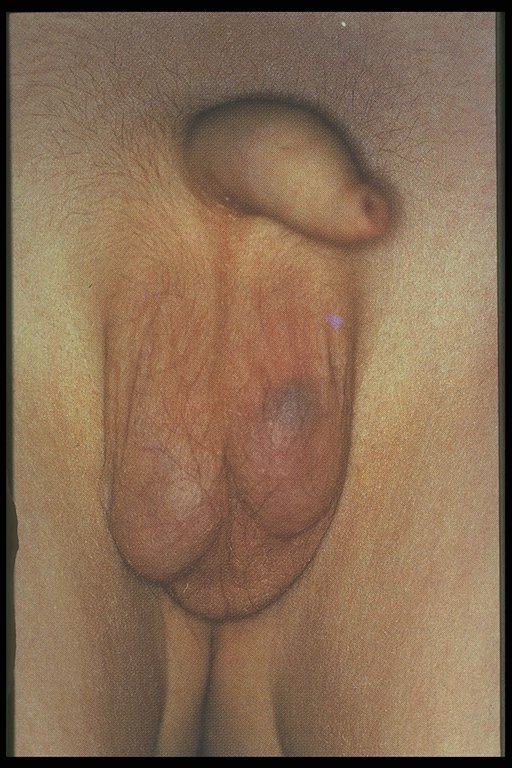 |
| Blue-dot sign |
 |
| Acute scrotum in an infant. |
Investigations
***DO NOT LET DELAY IN OBTAINING IMAGING INVESTIGATION DELAY SURGICAL INTERVENTION AND MANAGEMENT***
Ultrasound doppler has a sensitivity of 88% and specificity of 90%. Absence of blood flow to the affected testes is diagnostic. Parenchymal echonegicity is also decreased although it may be increased after infarction. Echogenic, enlarged epididymis is an ancillary sign of testicular torsion. Ultrasound may also help differentiate from hydrocele, abscess, hematoma or tumor.
Torsed appendix usually presents as hyper-echogenic nodule between the epididymis and testes.
Epididymitis may present as hypervascular epididymis although torsion should not be excluded based on just this feature.
Radio-isotope scan may also help in accurately diagnosing testicular torsion. This is unfortunately not widely available.
 |
| Note the absence of blood flow in the left testes. |
Management
***IF IN DOUBT OF DIAGNOSIS, SURGICALLY EXPLORE***
Manipulation of the testes has been attempted although most suggest that this is done only while waiting for preparation before surgical exploration. This is done in an 'open book' manner, rolling the testes laterally. Success of this maneuver usually relieves the pain spontaneously. This maneuver however is practically difficult to perform on a uncooperative child unless under anesthesia or adequate pain relief. Furthermore, if the torsion occurred in a lateral rotation, the maneuver may worsen the torsion.
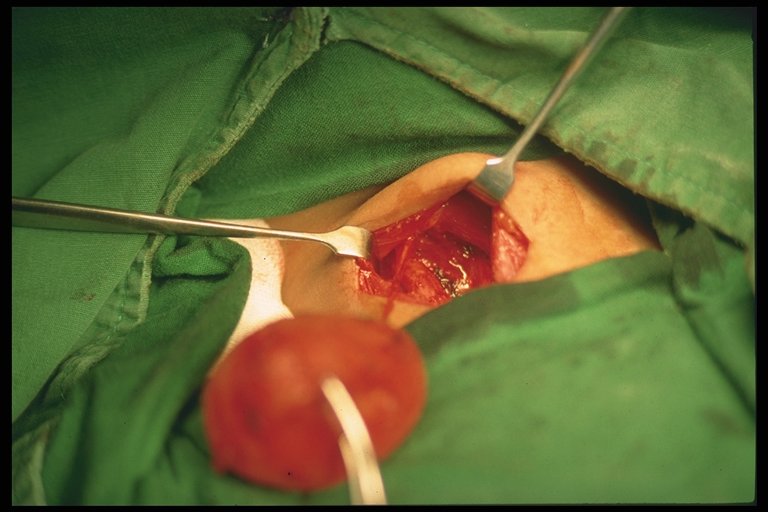
Surgical exploration is warranted if diagnosis is in doubt or there is high suspicion of testicular torsion. Incision may be made paramedian, transverse or vertically through the scrotum. The torsed tested is untwisted.
Examination is done to see testicular viability. If testes is infarcted, orchidopexy is recommended in patients more than 10 years old, as there is risk of autoimmunization with spermatogonia as there is a breech between the blood-testes barrier which may affect fertility at a later age even when the contralateral testes is uninvloved. Some surgeons leave the necrosed testes in-situ in children less than 10 years old.
If the testes is viable, the tested is fixed (orchidopexy). The contralateral testes is also fixed as well, especially when the torsion is associated with the bell-clapper anomaly as this usually occurs bilaterally. It is recommended to use non-absorbable sutures in fixation of the testes tunica albugenia and dartos layer. Use of absorbable sutures are associated with recurrent torsion.
 |
| Torsion of the testes. The testes is still viable. Incision of the testes with presence of bleeding may confirm this. |
In the torsed appendix of the testes, this is excised.
 |
| Necrosed torsioned testicular appendix. |
Surgical management is more controversial in torsion in the neonates.
Prognosis
Rate of testes salvage is inversely proportional with ischemia time. Testes is salvagable by 90% if exploration is performed within 4 - 6 hours after onset of symptoms, 50% if done at 12 hours, and 10% if present more than 24 hours.
Conclusion and Take-Homes
Testicular torsion is a surgical emergency which prompt exploration and treatment is important if the testes were to be salvaged. Diagnosis although may be made by imaging studies, should not cause delay in surgical exploration. The contralateral testes should be explored as well and orchidopexy performed with non-absorbable sutures.
References:
1.
Coran, Arnold G., et al., Pediatric Surgery, 7th Ed., Elsevier
2. Hutson, John M., et al., Jones' Clinical Pediatric Surgery Diagnosis and Management, 6th ed., Blackwell Publishing (2008).
3. Godbole, Prasad P., Testicular Problems in Children, Pediatric and Child Health, Vol 22:6, June 2012.
4. Govindarajan, Krishna K., Pediatric Testicular Torsion, Emedicine, Medscape article.
5. Various websites for the pictures in which none are mine.